单细胞转录组测序介绍
单细胞转录组测序(Single cell RNA sequencing),简单来说,就是在单个细胞水平上,对转录组水平进行测序分析的技术。传统测序方法通常在多细胞基础上进行,得到的是一堆细胞中信号的均值,丧失了细胞异质性(细胞之间的差异)的信息。而单细胞测序技术能够揭示单个细胞的基因表达状态,解决了传统测序无法解决的细胞异质性问题。
这项技术自2009年首次问世以来,持续不断地发展。近年来,单细胞测序经历了爆发式的发展和普及。它在肿瘤、发育生物学、微生物学、神经科学等领域发挥着重要作用,成为生命科学研究的焦点。单细胞转录组测序技术的不断发展将在精准治疗、生物医学研究和临床应用中发挥越来越重要的作用。单细胞测序技术正在推动生物学领域向前发展,为个性化预防和治疗提供了技术支持。
肿瘤研究:单细胞测序技术帮助揭示肿瘤内不同细胞类型的异质性,探索肿瘤微环境中细胞的转录组表达和免疫功能。
发育生物学:通过单细胞测序,我们可以追踪胚胎发育过程中不同细胞的基因表达变化,理解细胞分化和发育。
神经科学:研究单个神经元的转录组可以帮助我们理解神经系统的功能、神经退行性疾病以及神经元之间的相互作用。
免疫学:单细胞测序技术有助于研究免疫细胞的异质性,例如T细胞、B细胞和巨噬细胞的功能状态。
微生物学:通过单细胞测序,我们可以深入了解微生物群落中不同细菌、病毒和其他微生物的基因表达。
10x 3’单细胞
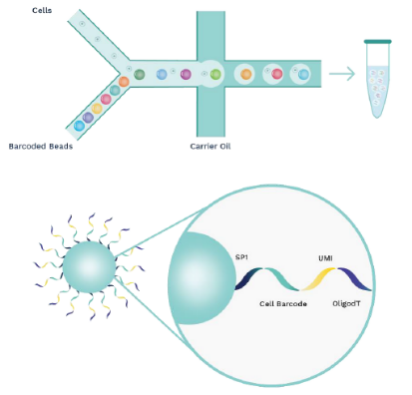
利用10x Genomics Chromium微流控系统,实现通过序列标签区别不同细胞和转录本。微流控系统通道直径很小一次通过一个细胞。首先在通道中加入带有标签的凝胶微珠,单个细胞和单个带有标签的凝胶微珠结合,二者被加入的油包裹到一个液滴中,形成油包水体系。凝胶微珠上带有大量包含凝胶微珠特异性Barcode序列、分子特异性UMI序列和polyT序列的引物序列,将细胞裂解后mRNA分子被引物序列中的polyT捕获,扩增后形成携带有Barcode和UMI序列的cDNA,即可实现将不同细胞来源的不同mRNA分子加上标签。
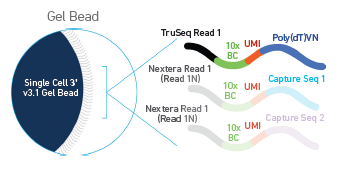
Gel beads在转录组测序中的作用是捕获细胞中的mRNA,每个微珠表面携带有几十万个Oligo序列(如下图)。Oligo dT序列由4个部分组成:read1用于上机测序;第二段是16bp 的10x barcode序列,每一个Gel Beads都携带不同的barcode , 可标记获取的mRNA都来自于哪一个细胞;第三段UMI长度为12bp, 可以将转录本序列进行绝对定量,避免扩增产生偏好;第四段是30bp的ployT,用于捕获有ployA尾的转录本。
对制备好的单细胞悬液先进行质检,质检合格后与Gel Beads和油分别加入到Chromium Chip G的不同通道,经由微流体“T字”交叉系统形成GEM。接着细胞裂解,Gel Beads自动溶解释放大量引物序列,与带有PolyA的mRNA进行逆转录,生成带有10x Barcode和UMI信息的cDNA第一链。
样本起始量与送样建议
|
组织类型 |
新鲜组织、新鲜悬液或速冻组织 |
|
细胞大小 |
<40 μm |
|
细胞活性 |
>85%(尽量去除细胞团/碎片/杂质等) |
|
细胞浓度 |
700~1200 cells/μL |
|
细胞总量 |
>5万 |
注意事项:详细样本准备指南,请联系在线客服。
生物信息学分析内容
 代表性文章
代表性文章
【1】Zheng H , Pomyen Y , Hernandez M O , et al. Single cell analysis reveals cancer stem cell heterogeneity in hepatocellular carcinoma[J]. Hepatology, 2018.
【2】Park J , Shrestha R , Qiu C , et al. Single-cell transcriptomics of the mouse kidney reveals potential cellular targets of kidney disease[J]. Science, 2018:eaar2131.
案例展示
Hepatology:单细胞分析显示肝癌干细胞的异质性
论文标题:Single-cell analysis reveals cancer stem cell heterogeneity in hepatocellular carcinoma
刊登日期:2018年7月
发表杂志:Hepatology
影响因子:14.079
研究机构:美国国家卫生研究院国家癌症研究所、曼谷朱拉蓬研究所、弗雷德里克国家癌症研究实验室
样本类型:肝癌细胞系(HuH1和HuH7) ,手术切除肿瘤样本P1
技术手段:单细胞测序(10X Chromium、SMART-seq)、流式分选
研究背景
肝细胞癌(HCC,hepatocellular carcinoma)的肿瘤内分子异质性部分归因于肝癌干细胞(CSC,hepatic cancer stem cells)的存在。 由各种细胞表面标志物定义的不同CSC群体可能包含不同的致癌驱动因素,这在定义靶向疗法时构成了挑战。 研究人员在单细胞水平(10X Chromium、SMART-seq)上整合转录组学和功能分析用以评估CSC的异质性程度。结果表明,CSC在单细胞水平上的表型、功能和转录均存在异质性性,不同的CSC亚群具有不同的分子特征。而且有趣的是,不同CSC亚群内的不同基因与HCC预后相关,并且彼此相互独立,表明CSC异质性影响肿瘤内异质性和肿瘤进展。
文章结果
大量数据表明CD13、CD24、CD44、CD90、CD133、EpCAM等表面标志物可以定义CSC,研究人员选择CD24、CD133、EpCAM分选HuH1和HuH7不同细胞亚群,用于研究CSC生物学和分子水平异质性。免疫荧光染色表明,在两种细胞培养形态下(单层培养、球体培养),两个HCC细胞系均表现出明显的细胞异质性。
HuH1和HuH7细胞系(原始细胞数分别是1343、1134)正常培养和缺氧培养(促进细胞干性)2周后,研究人员依照三种表面标志物(CD24、CD133、EpCAM)分选细胞,HuH1和HuH7中标志物阳性的CSCs比例分别为15-20%、8-12%,而标志物阴性CSCs比例只有0-5%(下图A-B),培养后不同细胞亚群的自我更新能力差异很大(下图C-D); 分选后的细胞经过1个月培养后,和未分的得细胞相比,CD133+、EpCAM+、CD24+、Triple+细胞能够扩大成为混合CSCs,而存活下的Triple-细胞能够扩大成混合CSCs,可能是由于部分细胞不是真正的标记阴性细胞,或者标记阴性细胞经历了转换变为标记阳性细胞(下图E)。
研究人员采用SMART-Seq方式分析HuH1和HuH7阳性标记细胞以及分选的P1(分选CD133+/CD24+/EpCAM+/CD45-细胞)细胞表达谱(单个细胞测序、10细胞混合测序、100细胞混合测序)信息,并与数据库中人胚胎干细胞(hESC)、鼠内皮细胞(mEC)和鼠造血干细胞(mHSC)表达谱数据做了比较,发现不同细胞类群具有不同的基因表达模式,且不同类型细胞中每个细胞的基因表达模式均存在差异,HuH7细胞间差异大于HuH1;当细胞混合(10细胞混合、100细胞混合)后这种差异程度明显降低(下图A)。而当单独聚类HuH7细胞时,发现不同标记细胞彼此可以区分(下图B)。以上数据均表明单细胞水平上转录组存在异质性。
研究人员比较了HuH7阳性标记细胞(Triple+)和阴性标记细胞(Triple-)基因表达差异,鉴定到的286个Triple+相关基因可以预测来自LCI队列的240例肿瘤样本、来自TCGA的250例肿瘤样本的整体生存率,但在非肿瘤样本中无意义。
不同标记CSC细胞群体基因表达比较分析,EpCAM+、CD133+、CD24+、Triple+特征基因基本不重合(下图A),IPA通路分析也表明不同细胞群体的通路也不重合(下图C-F),而且不同细胞群体特征基因可以用于生存期预测,且EpCAM+、CD133+比CD24+更有优势(下图B)。
为了整体了解细胞群体多样性,研究人员采用10X 单细胞测序方式,对3847个HuH1、HuH7、P1细胞进行了分析, HuH1可以分为2个亚群,HuH7分为4个细胞亚群,P1分为3个细胞亚群,细胞聚类结果与与SMART-seq结果类似(下图A)。CSC标志物,例如EpCAM、CD133 (PROM1)、CK-19 (KRT19) 和乙醛脱氢酶 (ALDH1A1) (下图B)以及HCC特异基因,AFP、MDK、GPC3、GOLM1、YY1AP1、PLK1, ECT2、NELFE(下图C)在不同细胞亚群中表达存在异质性。P1的3个细胞亚群中表达了山中因子 (KLF4、POU5F1、MYC), 干细胞marker (NANOG),白细胞marker(PTPRC), T细胞 markers (CD3E, CD8A) 和B细胞marker (CD79A) ,但其表达也存在异质性,其中第一个和第二个P1亚群为HCC浸润白细胞(分别表达T细胞marker和B细胞marker),第三个P1亚群主要为HCC细胞(下图D)。

参考文献
Hongping Zheng,et al. Single cell analysis reveals cancer stem cell heterogeneity in hepatocellular carcinoma. Hepatology, 2018, doi:10.1002hep.29778
结果展示
1. 单细胞亚群分类
针对PCA结果中选取前10个主成分,采用graph-based聚类方法对细胞进行分类。使用相同的数据进行t-SNE分析,然后将聚类结果在t-SNE映射中展示。
2. Marker基因分析
细胞群体Top10 Marker基因的小提琴图和表达映射图
3. 差异基因KEGG富集性分析
Pathway通路图展示与说明:红色代表注释到某个ko节点且上调的显著差异表达转录本,蓝色代表注释到某个ko节点且下调的显著差异表达转录本,橘黄色代表注释到某个ko节点不仅有上调又有下调的显著差异表达转录本。方框内的4位数字表示各种酶的EC编号;空心圆圈表示小分子化合物;实心箭头表示生化反应的方向;虚线箭头连接其他的相关代谢途径。下图仅为报告中的展示图片
常见问题(FAQ)
1、单细胞转录组测序需要做重复吗?
单细胞测序的目的在于鉴定细胞群体中的不同亚群、得到新的重要分子标志物和鉴定稀有细胞亚群等,因此更多的是关注细胞和细胞之间的差别。从目前已有的文献看,对重复数没有强制要求。但从近期的发文趋势来看,建议每组设置样本数3~5 个,临床样本根据实际情况建议每组 5 例以上。
2、组织样本如何制备单细胞悬液?
单细胞转录组测序目前没有一个统一的样本制备方法,针对于不同的组织样本,有不同类型的样本单细胞制备方法。此外,可以具体根据老师提供的具体样本类型,查阅相关的文献,探讨适合于老师样本的单细胞悬液制备方法。
3、单细胞转录组每个细胞能捕获到多少个基因?
根据不同的细胞类型,捕获到的基因数会有区别,有些细胞类型高的会在3000左右,而有些大概在1000左右。基因检出数与样本状态有关,不同类型的样本,基因检出数可能存在数倍的差异。
SeekOneDD 3’单细胞
SeekOneDD (Digital Droplet) 单细胞3’转录组测序采用SeekOneMM单细胞转录组试剂盒进行单细胞转录组建库,通过微孔芯片(SeekOneChip)和核酸标记磁珠(Barcoded Beads)即可实现单细胞的捕获、标记。最终构建出兼容Illumina 测序仪的高通量单细胞转录组文库进行测序,随后进行一系列生信分析。
SeekOneDD系统基于微流控技术原理,通过油包水实现单细胞分离捕获,利用凝胶微珠上Barcoded Beads对不同细胞进行标记,通过UMI的标签对mRNA进行定量,来构建高通量单细胞文库,数据下机后使用SeekOne Tools完成单细胞数据质控处理。
BD 3’单细胞
BD Rhapsody的技术不再采用利用微流控孔道射出细胞和射出的磁珠碰撞进行单细胞捕获,而是采用CytoSeq特有的蜂窝板技术。该技术用20W+的微孔(远大于Input细胞数量),保证单孔中的单细胞捕获。在细胞捕获完成后,进行细胞裂解,细胞释放出的mRNA可以被polydT的beads在微孔中捕获。
BD Rhapsody Beads
由以下3部分组成:
1. CL(细胞标签,cell label):一个磁珠上的CL序列相同,不同磁珠上的CL序列不同,从而达到区分细胞的作用;
2. UMI(unique molecular identifier):一个磁珠上的引物UMI各不相同,用于区分不同的mRNA分子,避免PCR偏好性;
3. ploy dT:与mRNA 3‘端poly A互补配对,以捕获polyA尾的mRNA;
BD Rhapsody是利用铺满20万个直径50 μm微孔的微孔板和携带细胞标签(cell label)的直径35 μm磁性beads捕获细胞。将制备好的单细胞悬液铺至微孔板上,细胞靠自身重力沉降至微孔底部,沉降速度慢的死细胞和杂质在这一步会被冲洗掉;控制细胞数目,减少两个细胞落入一个微孔的现象(形成doublets);接着将beads铺至微孔板,同样沉降至微孔板底部;冲洗掉多余的beads并加入细胞裂解液,细胞释放出的mRNA可以被polydT的beads在微孔中捕获。利用磁铁回收beads到单管中进行后续逆转录、建库等操作。
10x 5’单细胞及免疫组库
10x V(D)J 采用TCR/BCR恒定区的连续巢式引物对全长cDNA进行扩增,以获取个体中所有T/B细胞的BCR和TCR的V(D)J全长序列总和。在肿瘤微环境、感染性疾病、器官移植后排斥、免疫治疗等研究领域中有着广泛的应用。
技术优势
可实现成对的重链和轻链(B细胞)或α和β链(T细胞)的全长测序
精细到单细胞水平,能获得大量单细胞的免疫组库数据
大规模单细胞VDJ+表达谱测序,单个细胞成本大大降低
代表性文章
ElhamAzizi, et al. Single-Cell Map of Diverse Immune Phenotypes in the Breast Tumor Microenvironment. Cell, 2018,DOI: https://doi.org/10.1016/j.cell.2018.05.060
案例展示
单细胞测序揭示复杂的肿瘤微环境
研究背景
了解肿瘤微环境中的免疫细胞表型,对了解癌症进展和免疫疗法的机制是必不可少的。本研究从8名乳腺癌患者的肿瘤样本及配对的正常血液、乳腺和淋巴结样本中获得多个免疫细胞,开展单细胞RNA测序实验。之后,利用SEQC流程和“Biscuit”算法,对这些组织中的免疫细胞进行聚类和鉴定。
研究思路
本研究总共鉴定出83个不同的免疫细胞簇,包括38个T细胞簇、27个骨髓谱系细胞簇、9个B细胞簇和9个自然杀伤细胞簇。大多数的细胞簇(除了其中的10个)是不止一名患者所共有的,尽管在肿瘤样本中的表型往往不同。研究指出,部分T细胞在肿瘤和正常组织中都存在,不过细胞毒性T细胞在肿瘤中更丰富,Treg细胞也是如此。
在一系列后续实验中,研究人员利用10X Genomics的方法获得27,000多个T细胞的TCR-seq和RNA-seq配对数据并开展分析。比较了正常组织和乳腺癌组织的免疫组库,发现表型扩增往往发生在乳腺癌微环境中。
这些观察结果,以及免疫细胞的RNA-seq和TCR-seq数据集和全面的分析平台,将有助于研究人员更好地了解免疫细胞促进和延缓肿瘤生长背后的分子机制。
参考文献
ElhamAzizi, et al. Single-Cell Map of Diverse Immune Phenotypes in the Breast Tumor Microenvironment. Cell, 2018,DOI: https://doi.org/10.1016/j.cell.2018.05.060
结果展示
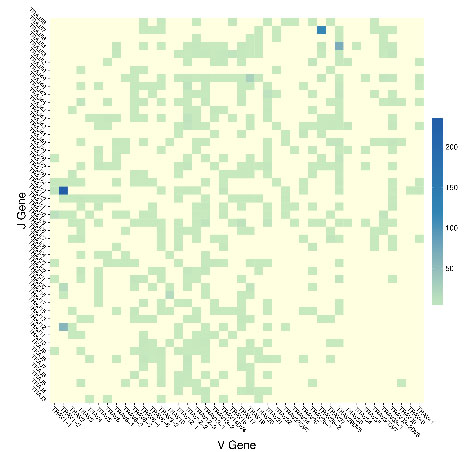
1.V-J paired特征分析
V-J paired直接反应出CDR3或免疫组库的变化,图中横坐标为V Gene,纵坐标为J Gene。颜色越深表示V-J paired丰度越高。
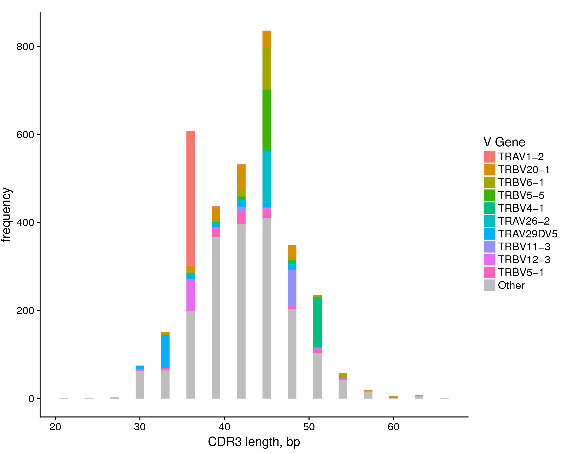
2.V/J基因特征分析
对CDR3 V/J基因序列长度分布情况进行统计,图中横坐标为CDR3(V Gene)碱基序列长度,纵坐标为对应的clonotype name频率,不同颜色表示不同V Gene,top10的V Gene用彩色表示,other(灰色部分)表示其他V Gene。
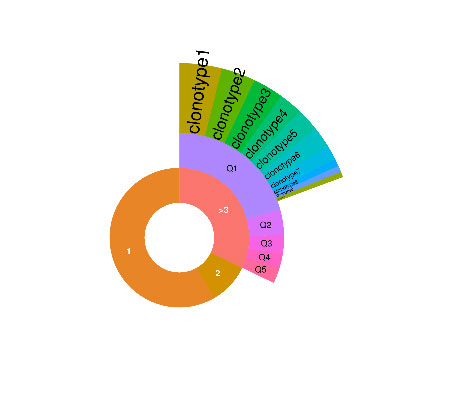
3.Clonotype分型
一个clonotype(克隆亚型)的定义为:一组唯一的CDR3核苷酸序列。Cell Ranger根据拼接得到的Consensus中的CDR3氨基酸序列进行Clonotype分型,其有效Barcode为该Clonotype的丰度。利用圈图展示免疫组库的top10 clonotype频率分布(clonality)。
10x 单细胞ATAC-seq
ATAC-seq(Assay for Transposase Accessible Chromatin with high-throughput sequencing,染色质易开放区域测序)是2013年由美国Stanford大学的William Greenleaf教授研发了一种全新的方法,利用DNA转座酶结合高通量测序技术,来研究染色体的可进入性。ATAC-seq的原理是,染色质易开放区域测序)利用Tn5转座酶切割染色质的开放区域,并加上测序引物进行高通量测序,通过生物信息分析鉴定转录因子结合位点和核小体区域位置,从而为研究基因调控、DNA 印记等提供有效的方法。通过研究特定状态下开放染色质,可以在DNA水平研究基因转录调控。
传统的寻找全基因组范围内开放的染色质区域采用的方式包括ATAC-seq、MNase-seq和DNase-seq,FAIRE-seq、chip-seq等。10x Genomics Chromium Single Cell ATAC Solution(10X scATAC-seq)是10 X Genomics新推出单细胞表观基因组学水平上揭示染色质可接近性的研究策略。通过单细胞染色质可接近性图谱构建,不仅能清晰了解染色质结构,还可以绘制高分辨率转录因子调控网络,为深入了解疾病的诊断和治疗铺平道路。
技术优势
可检测单细胞转录调控区域中的开放染色质
每个通道可容纳500-10000个细胞核
细胞核捕获率高达65%
运用10x官方软件和工具,可对单细胞基因调控网络进行精细化分析
代表性文章
Buenrostro J D , Corces M R , Lareau C A . Integrated Single-Cell Analysis Maps the Continuous Regulatory Landscape of Human Hematopoietic Differentiation.[J]. Cell, 2018.
案例展示
联合不同单细胞分析技术绘制人类造血系统分化的连续调节谱
研究背景
造血分化 是从造血干细胞分化为具有不同功能的细胞过程,在成人中主要发生在骨髓,最终产生淋巴系细胞(B,T,NK cell),髓系-粒系(monocyte, granulocyte),红系(Ery,mega)等细胞类型,是研究干细胞分化、肿瘤免疫、血液疾病等的良好模型。造血分化过程是一个复杂、多阶段的,受多种因子调控的过程,单细胞表观基因组分析有助于解析造血干细胞转录和细胞命运异质性的顺式和反式调节机制。
研究思路
首先利用scATAC-seq数据从chromatin accessibility入手,分析造血分化过程染色质开放状态图谱的异质性;用ChromVAR R包预测TF的活动;采用多种聚类方法,如通过TF Z scores利用Hierarchical clustering 和t-SNE聚类,并采用了reference guided approach,基于bulk sample和算法对单细胞数据进行降维聚类分析;整合scRNA-seq 和scATAC-seq数据分析TF与chromatin accessibility,以及顺式调控元件和邻近基因的表达的相互关系。
研究结果
研究人员通过单细胞ATAC-Seq的研究策略,分析了10类已定义的人类造血系统分化细胞类型中染色质可接近性的数据,构建造血细胞分化中染色质可接近性状态的变化轨迹;并整合单细胞转录组数据,构建了转录因子动态调控轨迹。
从单细胞RNA-seq和ATAC-seq的整合分析提供了对人类造血功能的调节特征和动态的见解,在实验或计算上整合多种单细胞数据类型的进一步改进将为发育或疾病细胞命运决策提供单细胞水平上的视角。期望未来的工作结合单细胞表观基因组学、转录组学、蛋白质组学和谱系测量可以揭示对启动调节多种细胞命运转换的调节因子的分子机制和时间顺序的重要见解。
参考文献
Buenrostro J D , Corces M R , Lareau C A . Integrated Single-Cell Analysis Maps the Continuous Regulatory Landscape of Human Hematopoietic Differentiation.[J]. Cell, 2018.
结果展示
1. 细胞聚类
Cell Ranger ATAC 利用每个细胞的peaks 信息,运行cluster mode 对这些细胞进行分群。经tSNE 降维算法,得到细胞聚类的二维结果。
2. 数据可视化
10X genomics公司提供的专门的分析结果查看软件-Loupe Cell Browser,该软件是一个图形界面的软件,操作非常的方便。可以用于以下应用:1.确定细胞类型: 使用可及性模式发现能够区分不同细胞类型和功能组别的启动子和转录因子motif;2.分析可及性差异: 使用ATAC Peak Viewer 定位具有假定调节功能的不同可访问基因组区域的位置;3.发现显著性特征: 创建自定义聚类并使用差异可及性分析工具精准识别数据中的细胞组别;4.共享数据结果: 保存感兴趣的信息,导出数据表,并获取单细胞ATAC数据的屏幕截图。