标记蛋白组TMT
TMT(Tandem Mass Tag)技术由美国Thermo Fisher Scientific开发的一种多肽体外标记技术。该技术采用了6标(TMTsixplex™ Isobaric Label Reagent Set)、10标(TMT10plex™ Isobaric Label Reagent Set )、16标(TMTpro 16plex)、18标(TMTpro 18plex)等同位素标签,通过与肽段特异氨基酸位点相连实现不同来源的肽段标记,然后进行串联质谱分析,监测碎裂下来的标签实现肽段定量。一次实验可灵活比较最多18种不同样本中蛋白质的相对含量。
TMT试剂主要由3部分组成:报告基团、平衡基团和肽反应基团。以6标试剂为例:报告基团相对分子质量分别为126、127、128、129、130和131;平衡基团相对分子质量分别为103、102、101、100、99和98,这样就形成了6种相对分子质量均为229的等量异位标签。报告离子的强度就代表了相应样品中蛋白质/肽段的强度,实现了对不同样本中蛋白质的定性和定量分析。
技术优势
1. 灵敏度高:低丰度蛋白也能检测出
2. 适用范围广:几乎可对任何物种的各类蛋白质进行分离鉴定
3. 高通量:能同时对18组样本中包含的蛋白进行鉴定及表达差异分析
4. 高效:液相色谱与串联质谱连用,自动化操作,分析速度快,分离效果好
样本起始量与送样建议
|
样本类型 |
起始量 |
|
动物及临床脏器组织/脑组织等 |
>20mg |
|
动物及临床皮肤/骨/血管/脂肪组织等 |
>100mg |
|
植物叶片组织/花 |
>200mg |
|
植物根/茎/果实/种子 |
>500mg |
|
原代细胞/细胞系 |
>5×106个 |
|
总RNA |
>100μg |
注意事项:详细样本准备指南,请联系在线客服。
生物信息学分析内容
 项目文章
项目文章
Luo Z, Peng W, Xu Y, Xie Y, Liu Y, Lu H, Cao Y, Hu J. Exosomal OTULIN from M2 macrophages promotes the recovery of spinal cord injuries via stimulating Wnt/β-catenin pathway-mediated vascular regeneration. Acta Biomater. 2021 Dec;136:519-532. PMID: 34551329.
常见问题
1. iTRAQ及TMT等标记定量蛋白质组技术有什么特点?
iTRAQ以及TMT等标记定量蛋白质组技术分辨率高,细胞样品最多可鉴定超过6000个蛋白,并且绝大多数蛋白都有定量和定性信息;其次是通量高,可以一次最多完成8个(iTRAQ)或者10个(TMT)样品的实验,特别适合于多组样品间的同时比较以及生物学过程的动态检测。
2. 没有全基因组数据的物种该如何进行蛋白的鉴定?
对于已经公布全基因组序列的物种,可以在得到全序列后构建本地建库并进行本地检索;对于未进行全基因组测序的物种,可以采用近缘物种的数据库或者EST数据以及转录组数据进行检索。
4D-DIA
DIA主要采用数据非依赖采集模式(DIA),结合传统的数据依赖采集模式(DDA)构建参考谱图库,通过液质联用技术(LC-MS/MS)将质谱整个全扫描范围分为若干个窗口,高速、循环地对每个窗口中的所有离子进行选择、碎裂、检测,从而无遗漏、无差异地获得样本中所有离子的全部碎片信息。随后利用搜库软件进行蛋白组学的鉴定、定量分析等。
技术优势
灵敏度高,无歧视地获得所有肽段的信息,不会造成低丰度蛋白信息的丢失,
循环时间固定,扫描点数均匀,定量准确度高,
重复性好,重复样品间的定量相关性可达到0.99以上。
分析内容
1. 数据质控
2. 蛋白功能注释:GO 注释、COG 注释、KEGG 注释、结构域注释
3.蛋白定量分析:蛋白定量结果、蛋白表达水平聚类分析、重复性分析
4. 蛋白差异分析:蛋白差异分析结果、差异蛋白火山图、差异蛋白聚类热图
5. GO 富集分析:GO 富集结果、GO 富集柱状图、GO 富集有向无环图、
6. KEGG 富集分析:KEGG 富集结果、KEGG 富集气泡图、KEGG 富集通路图
7. 结构域富集分析:结构域富集结果、结构域富集柱状图、差异蛋白互作分析、
8. 差异蛋白互作分析
9. 转录组关联分析
代表性文章
【1】Collins B C , Hunter C L , Liu Y , et al. Multi-laboratory assessment of reproducibility, qualitative and quantitative performance of SWATH-mass spectrometry[J]. Nature Communications, 2017, 8(1):291.
【2】Rouwette T , Sondermann J , Avenali L , et al. Standardized profiling of the membrane-enriched proteome of mouse dorsal root ganglia provides novel insights into chronic pain[J]. Molecular & Cellular Proteomics, 2016, 15(6):mcp.M116.058966.
案例展示
蛋白定量DIA用于慢性疼痛的研究
Standardized Profiling of The Membrane-Enriched Proteome of Mouse Dorsal Root Ganglia (DRG) Provides Novel Insights Into Chronic Pain
研究背景
慢性疼痛是一种治疗方案有限的复杂疾病。虽对其发病机理进行了多次研究,但各结果间存在较大不一致性,主要受限于传统蛋白质组shotgun技术固有的缺陷。发病机制依然不清晰。
实验设计
炎症性疼痛和神经性疼痛两种模型鼠及其对应control鼠共4组,各3个生物学重复
每个生物学重复由10-13只鼠pooling而成
解剖获得同侧背根神经节膜部分提取蛋白进行DIA蛋白定量

图1 实验设计类型
主要发现
1)DDA建库鉴定到3067个蛋白,迄今鉴定数最多的背根神经节蛋白研究。
2)DIA四组都鉴定到的蛋白有2526个,其中CFA和Vehicle都鉴定到的有2581个;SNI和Sham都鉴定到的2600个,表明实验重复性较好。
3)炎症性和神经性疼痛差异蛋白分别为64和77,两种模式共有差异蛋白为12个。
4)其中Serca蛋白在两种模式小鼠中表达量变化相反,表明两种疼痛的调控机制不同。
5)western blot和免疫组化对上述DIA结果进行了验证。
图2 四组实验模型的蛋白聚类分析
参考文献
Rouwette T , Sondermann J , Avenali L , et al. Standardized profiling of the membrane-enriched proteome of mouse dorsal root ganglia provides novel insights into chronic pain[J]. Molecular & Cellular Proteomics, 2016, 15(6):mcp.M116.058966.
结果展示
结构域注释柱状图
Interproscan是蛋白质结构域和功能注释最常用的软件之一。为了能更全面的进行结构域的注释,Interproscan整合了一些最常用的结构域数据库,包括 Pfam、 ProDom、 SMART 等结构域的数据库,利用模式结构或特征进行功能未知蛋白的结构域注释,绘制结构域注释的柱状图。横坐标代表蛋白数目,纵坐标代表注释到的 IPR 条目。
蛋白表达水平聚类图
蛋白表达水平聚类分析用于判断不同实验条件下蛋白表达量的相关性。每个样品都会得到一个绝对或相对蛋白表达集合,将所有样品表达集合并在一起,用于层次聚类分析和 K-means 聚类分析。
KEGG 富集通路图
在 KEGG 通路图中,圈出了差异蛋白,其中绿色底色框内为鉴定出的总蛋白,蓝色框标记的是下调差异蛋白,红色框标记的是上调差异蛋白,黄色框标记的是通路中该功能对应的多个蛋白中既有上调差异蛋白、也有下调差异蛋白。
差异蛋白互作分析
利用 StringDB 蛋白质互作数据库进行鉴定蛋白的互作分析,若在数据库中有相应的物种,则直接提取相应物种的序列,若无,则提取近源物种的序列,然后将差异蛋白的序列与提取出的序列进行 blast 比对,得出相应的互作信息,构建网络图。
常见问题
1. 蛋白定量DIA和传统的非标记定量有何优势?
传统的非标记定量一般需要通过分级的方法开展,耗时久、重复性差、数据结果可信度不高;蛋白定量DIA技术是新一代非标记定量技术,可通过更优的数据采集模式,在相同时间采集到更丰富的信息,压缩周期的同时,大福度提高样本间平行比较的重复性,数据结果可信度极高。
DirectDIA
DirectDIA(direct Data-independent acquisition),即DDA-free DIA(dDIA),与传统DIA分析策略相比,不用再进行DDA分级建库,利用机器深度学习实现直接通过搜索DIA原始文件谱图生成库。深度学习打分寻找谱峰碎裂规律,预测保留时间,去除假阳性结果。改进后的这种方式提高了效率,降低了成本,保留了DIA可重复的定量的优势,鉴定到的蛋白数目与传统DIA的差距也越来越小。
修饰蛋白组
蛋白质翻译后修饰 (Protein translational modifications,PTMs) 通过功能基团或蛋白质的共价添加、调节亚基的蛋白水解切割或整个蛋白质的降解来增加蛋白质组功能多样性。这些修饰几乎影响正常细胞生物学和发病机制的所有方面。因此,识别和理解PTM在细胞生物学和疾病治疗和预防的研究中至关重要。由于存在大量不同的PTM,无法对所有可能的蛋白质修饰进行全面介绍,因此仅对当今蛋白质研究中研究的最常见PTM,如磷酸化、乙酰化、泛素化、糖基化等,进行概述:
(1)磷酸化:可逆的蛋白质磷酸化,主要发生在丝氨酸、苏氨酸或酪氨酸残基上,是目前研究得最深入的翻译后修饰之一。磷酸化在细胞周期、生长、凋亡和信号转导等过程中起着重要的调控作用。
(2)乙酰化:几乎所有的真核细胞蛋白质都通过不可逆和可逆的机制发生 N-乙酰化,或乙酰基转移到氮上。N 端乙酰化需要 N 端蛋氨酸被蛋氨酸氨基肽酶 (MAP) 裂解,然后用 N-乙酰基转移酶 (NAT) 用乙酰辅酶 a 的乙酰基取代氨基酸。这种类型的乙酰化是共翻译的,因为 N 端在仍与核糖体相连的生长中的多肽链上乙酰化。
(3)泛素化:泛素是一类分子量为 8 kDa 的多肽,由 76 个氨基酸组成,通过泛素 C 末端的甘氨酸连接到靶蛋白赖氨酸的 ε-NH2 上。在最初的单泛素化事件之后,可能形成泛素聚合物,然后 26S 蛋白酶体识别多聚泛素化蛋白,26S 蛋白酶体催化泛素化蛋白的降解和泛素的再循环。
(4)糖基化:蛋白质糖基化是蛋白质翻译后的主要修饰之一,对蛋白质的折叠、构象、分布、稳定性和活性都有重要影响。
PRM靶向蛋白组
PRM(平行反应监测,Parallel Reaction Monitoring),是目前靶向蛋白质组学数据采集的主流方法,通过对特异性肽段或目标肽段(如发生翻译后修饰的肽段)进行选择性检测,从而实现对目标蛋白质/修饰肽段的靶向相对或绝对定量。PRM可用于组学数据验证,比如基因组/转录组/定量蛋白质组/修饰蛋白质组获得的目标基因/蛋白,实现其相对/绝对定量分析。
Olink靶向蛋白组
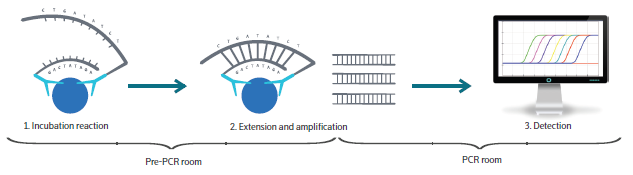
Olink蛋白质组学技术是一种高通量的蛋白质检测方法,可以同时检测数百种蛋白质,并且具有高灵敏度和高特异性。其技术原理主要是:
Probe 设计:Olink技术使用了一种称为 proximity extension assay(PEA)的方法,利用两种具有特异性的抗体,即Proximity Probe,分别识别待检测蛋白质的不同位点。这两种Proximity Probe 上分别连接有一种DNA分子,当两种Probe接近并结合到待测蛋白上时,两种DNA分子可以形成一个连通的DNA链。
扩增:所有DNA链同时被扩增成数百万个复制物,并且在扩增过程中添加了标记的核酸,这些标记的核酸是与每个DNA链上的特定序列相匹配的。
检测:使用Universal PCR Probe探针与所有扩增的DNA序列相匹配,PCR反应产生荧光信号,标记的核酸序列数量与待检测蛋白质的数量成正比。
 技术服务优势
技术服务优势
1. 高通量:快速检测批量样品中的48-3072种蛋白标志物;
2. 低体积:每个样品仅需不到1-6微升;
3. 靶向性:所有检测蛋白,均为生物标志物,覆盖100% 信号通路;
4. 高灵敏及宽动态:检测灵敏度可达fg/ml级别;动态范围横跨10个log,覆盖高、中、低丰度蛋白;
5. 自动化:自动化样品处理流程提供了绝佳的简易性、准确性和重复性。







